
Artificial intelligence in COVID-19 drug repurposing
Drug repurposing or repositioning is a technique whereby existing drugs are used to treat emerging and challenging diseases, including COVID-19. Drug repurposing has become a promising approach because of the opportunity for reduced development timelines and overall costs. In the big data era, artificial intelligence (AI) and network medicine offer cutting-edge application of information science to defining disease, medicine, therapeutics, and identifying targets with the least error. In this Review, we introduce guidelines on how to use AI for accelerating drug repurposing or repositioning, for which AI approaches are not just formidable but are also necessary. We discuss how to use AI models in precision medicine, and as an example, how AI models can accelerate COVID-19 drug repurposing. Rapidly developing, powerful, and innovative AI and network medicine technologies can expedite therapeutic development. This Review provides a strong rationale for using AI-based assistive tools for drug repurposing medications for human disease, including during the COVID-19 pandemic.

Network Medicine Framework for Identifying Drug Repurposing Opportunities for COVID-19
Coronavirus Disease-2019 (COVID-19) is an infectious disease caused by the SARS-CoV-2 virus. Various studies exist about the molecular mechanisms of viral infection. However, such information is spread across many publications and it is very time-consuming to integrate, and exploit. We develop CoVex, an interactive online platform for SARS-CoV-2 host interactome exploration and drug (target) identification. CoVex integrates virus-human protein interactions, human protein-protein interactions, and drug-target interactions. It allows visual exploration of the virus-host interactome and implements systems medicine algorithms for network-based prediction of drug candidates. Thus, CoVex is a resource to understand molecular mechanisms of pathogenicity and to prioritize candidate therapeutics. We investigate recent hypotheses on a systems biology level to explore mechanistic virus life cycle drivers, and to extract drug repurposing candidates. CoVex renders COVID-19 drug research systems-medicine-ready by giving the scientific community direct access to network medicine algorithms. It is available at https://exbio.wzw.tum.de/covex/

Exploring the SARS-CoV-2 virus-host-drug interactome for drug repurposing
Coronavirus Disease-2019 (COVID-19) is an infectious disease caused by the SARS-CoV-2 virus. Various studies exist about the molecular mechanisms of viral infection. However, such information is spread across many publications and it is very time-consuming to integrate, and exploit. We develop CoVex, an interactive online platform for SARS-CoV-2 host interactome exploration and drug (target) identification. CoVex integrates virus-human protein interactions, human protein-protein interactions, and drug-target interactions. It allows visual exploration of the virus-host interactome and implements systems medicine algorithms for network-based prediction of drug candidates. Thus, CoVex is a resource to understand molecular mechanisms of pathogenicity and to prioritize candidate therapeutics. We investigate recent hypotheses on a systems biology level to explore mechanistic virus life cycle drivers, and to extract drug repurposing candidates. CoVex renders COVID-19 drug research systems-medicine-ready by giving the scientific community direct access to network medicine algorithms.
A Network Medicine Approach to Investigation and Population-based Validation of Disease Manifestations and Drug Repurposing for COVID-19
The global Coronavirus Disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has led to unprecedented social and economic consequences. The risk of morbidity and mortality due to COVID-19 increases dramatically in the presence of co-existing medical conditions while the underlying mechanisms remain unclear. Furthermore, there are no proven effective therapies for COVID-19. This study aims to identify SARS-CoV-2 pathogenesis, diseases manifestations, and COVID-19 therapies using network medicine methodologies along with clinical and multi-omics observations. We incorporate SARS-CoV-2 virus-host protein-protein interactions, transcriptomics, and proteomics into the human interactome.

Multi-level proteomics reveals host-perturbation strategies of SARS-CoV-2 and SARS-CoV
The sudden global emergence of SARS-CoV-2 urgently requires an in-depth understanding of molecular functions of viral proteins and their interactions with the host proteome. Several omics studies have extended our knowledge of COVID-19 pathophysiology, including some focused on proteomic aspects1–3. To understand how SARS-CoV-2 and related coronaviruses manipulate the host we here characterized interactome, proteome and signaling processes in a systems-wide manner. This identified connections between the corresponding cellular events, revealed functional effects of the individual viral proteins and put these findings into the context of host signaling pathways. We investigated the closely related SARS-CoV-2 and SARS-CoV viruses as well as the influence of SARS-CoV-2 on transcriptome, proteome, ubiquitinome and phosphoproteome of a lung-derived human cell line. Projecting these data onto the global network of cellular interactions revealed relationships between the perturbations taking place upon SARS-CoV-2 infection at different layers and identified unique and common molecular mechanisms of SARS coronaviruses. The results highlight the functionality of individual proteins as well as vulnerability hotspots of SARS-CoV-2, which we targeted with clinically approved drugs. We exemplify this by identification of kinase inhibitors as well as MMPase inhibitors with significant antiviral effects against SARS-CoV-2.
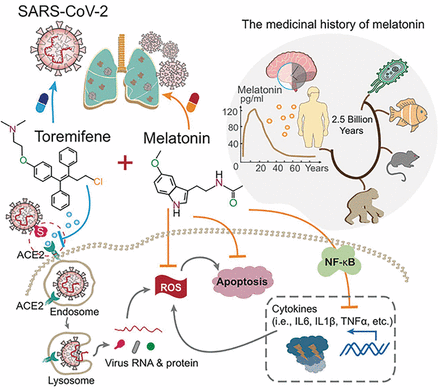
COVID-19 treatment: Combining anti-inflammatory and antiviral therapeutics using a network-based approach
To date, there are no effective antiviral medications for COVID-19. Drug repurposing, a strategy that uses existing drugs, offers potential prevention and treatment options for COVID-19. We discuss one treatment strategy that combines anti-inflammatory (melatonin) and antiviral (toremifene) agents for patients infected with SARS-CoV-2 from network medicine-based findings. We also describe the pathobiology and immunologic characteristics of COVID-19 and highlight the rationale of combination drug treatment to rescue the pulmonary and cardiovascular conditions resulting from COVID-19. A preliminary analysis reveals a high potential for the synergistic effects of melatonin and toremifene to reduce viral infection and replication, and the aberrant host inflammatory responses, offering strong biologic plausibility as an effective therapy for COVID-19.

Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2
We have previously provided the first genetic evidence that angiotensin converting enzyme 2 (ACE2) is the critical receptor for severe acute respiratory syndrome coronavirus (SARS-CoV), and ACE2 protects the lung from injury, providing a molecular explanation for the severe lung failure and death due to SARS-CoV infections. ACE2 has now also been identified as a key receptor for SARS-CoV-2 infections, and it has been proposed that inhibiting this interaction might be used in treating patients with COVID-19. However, it is not known whether human recombinant soluble ACE2 (hrsACE2) blocks growth of SARS-CoV-2. Here, we show that clinical grade hrsACE2 reduced SARS-CoV-2 recovery from Vero cells by a factor of 1,000–5,000. An equivalent mouse rsACE2 had no effect. We also show that SARS-CoV-2 can directly infect engineered human blood vessel organoids and human kidney organoids, which can be inhibited by hrsACE2. These data demonstrate that hrsACE2 can significantly block early stages of SARS-CoV-2 infections.

Barabási Lab
In a proof of concept, a group of Network Medicine scientists / clinicians lead by Professors Albert-László Barabási and Joseph Loscalzo (founders of the field of network medicine) using Network Medicine tools. Identified the human tissues that express the proteins needed by the virus (those that are needed for a statistically significant viral module, following Kitsak et al.). These are the tissues/organs that could be invaded by the virus. The insights that are evolving from these lines of studies are establishing preliminary predictive pipelines of distinct drugs that can be repurposed and applied to the SARS-CoV-2 infection and treatment of COVID-19.

Journal pre-proof
We have previously provided the first genetic evidence that Angiotensin converting enzyme 2 (ACE2) is the critical receptor for SARS-CoV and that ACE2 protects the lung from injury, providing a molecular explanation for the severe lung failure and death due to SARS-CoV infections. ACE2 has now also been identified as a key receptor for SARS-CoV-2 infections and it has been proposed that inhibiting this interaction might be used in treating patients with COVID19. However, it is not known whether human recombinant soluble ACE2 (hrsACE2) blocks growth of SARS-CoV-2. Here we show that clinical grade hrsACE2 reduced SARS-CoV-2 recovery from Vero cells by a factor of 1,000-5,000. An equivalent mouse rsACE2 had no effect. We also show that SARS-CoV-2 can directly infect engineered human blood vessel organoids and human kidney organoids, which can be inhibited by hrsACE2. These data demonstrate that hrsACE2 can significantly block early stages of SARS-CoV-2 infections.
A SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug-Repurposing
An outbreak of the novel coronavirus SARS-CoV-2, the causative agent of COVID-19 respiratory disease, has infected over 290,000 people since the end of 2019, killed over 12,000, and caused worldwide social and economic disruption1,2. There are currently no antiviral drugs with proven efficacy nor are there vaccines for its prevention. Unfortunately, the scientific community has little knowledge of the molecular details of SARS-CoV-2 infection. To illuminate this, we cloned, tagged and expressed 26 of the 29 viral proteins in human cells and identified the human proteins physically associated with each using affinity- purification mass spectrometry (AP-MS), which identified 332 high confidence SARS-CoV-2-human protein-protein interactions (PPIs). Among these, we identify 66 druggable human proteins or host factors targeted by 69 existing FDA-approved drugs, drugs in clinical trials and/or preclinical compounds, that we are currently evaluating for efficacy in live SARS-CoV-2 infection assays. The identification of host dependency factors mediating virus infection may provide key insights into effective molecular targets for developing broadly acting antiviral therapeutics against SARS-CoV-2 and other deadly coronavirus strains.

Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2
Human coronaviruses (HCoVs), including severe acute respiratory syndrome coronavirus (SARS-CoV) and 2019 novel coronavirus (2019-nCoV, also known as SARS-CoV-2), lead global epidemics with high morbidity and mortality. However, there are currently no effective drugs targeting 2019-nCoV/SARS-CoV-2.

Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2
Human coronaviruses (HCoVs), including severe acute respiratory syndrome coronavirus (SARS-CoV) and 2019 novel coronavirus (2019-nCoV, also known as SARS-CoV-2), lead global epidemics with high morbidity and mortality. However, there are currently no effective drugs targeting 2019-nCoV/SARS-CoV-2. Drug repurposing, representing as an effective drug discovery strategy from existing drugs, could shorten the time and reduce the cost compared to de novo drug discovery. In this study, we present an integrative, antiviral drug repurposing methodology implementing a systems pharmacology-based network medicine platform, quantifying the interplay between the HCoV–host interactome and drug targets in the human protein–protein interaction network. Phylogenetic analyses of 15 HCoV whole genomes reveal that 2019-nCoV/SARS-CoV-2 shares the highest nucleotide sequence identity with SARS-CoV (79.7%). Specifically, the envelope and nucleocapsid proteins of 2019-nCoV/SARS-CoV-2 are two evolutionarily conserved regions, having the sequence identities of 96% and 89.6%, respectively, compared to SARS-CoV. Using network proximity analyses of drug targets and HCoV–host interactions in the human interactome, we prioritize 16 potential anti-HCoV repurposable drugs (e.g., melatonin, mercaptopurine, and sirolimus) that are further validated by enrichment analyses of drug-gene signatures and HCoV-induced transcriptomics data in human cell lines. We further identify three potential drug combinations (e.g., sirolimus plus dactinomycin, mercaptopurine plus melatonin, and toremifene plus emodin) captured by the “Complementary Exposure” pattern: the targets of the drugs both hit the HCoV–host subnetwork, but target separate neighborhoods in the human interactome network. In summary, this study offers powerful network-based methodologies for rapid identification of candidate repurposable drugs and potential drug combinations targeting 2019-nCoV/SARS-CoV-2.

A genome-wide positioning systems network algorithm for in silico drug repurposing
Recent advances in DNA/RNA sequencing have made it possible to identify new targets rapidly and to repurpose approved drugs for treating heterogeneous diseases by the ‘precise’ targeting of individualized disease modules. In this study, we develop a Genome-wide Positioning Systems network (GPSnet) algorithm for drug repurposing by specifically targeting disease modules derived from individual patient’s DNA and RNA sequencing profiles mapped to the human protein-protein interactome network.

Network-based prediction of drug combinations
Drug combinations, offering increased therapeutic efficacy and reduced toxicity, play an important role in treating multiple complex diseases. Yet, our ability to identify and validate effective combinations is limited by a combinatorial explosion, driven by both the large number of drug pairs as well as dosage combinations. Here we propose a network-based methodology to identify clinically efficacious drug combinations for specific diseases. By quantifying the network-based relationship between drug targets and disease proteins in the human protein–protein interactome, we show the existence of six distinct classes of drug–drug–disease combinations. Relying on approved drug combinations for hypertension and cancer, we find that only one of the six classes correlates with therapeutic effects: if the targets of the drugs both hit disease module, but target separate neighborhoods. This finding allows us to identify and validate antihypertensive combinations, offering a generic, powerful network methodology to identify efficacious combination therapies in drug development.

Time-Resolved Systems Medicine Reveals Viral Infection-Modulating Host Targets
Drug-resistant infections are becoming increasingly frequent worldwide, causing hundreds of thousands of deaths annually. This is partly due to the very limited set of protein drug targets known for human-infecting viral genomes. The eleven influenza virus proteins, for instance, exploit host cell factors for replication and suppression of the antiviral immune responses. A systems medicine approach to identify relevant and druggable host factors would dramatically expand therapeutic options. Therapeutic target identification, however, has hitherto relied on static molecular networks, whereas in reality the interactome, in particular during an infection, is subject to constant change.

A structural transition in physical networks
In many physical networks, including neurons in the brain1,2, three-dimensional integrated circuits3 and underground hyphal networks4, the nodes and links are physical objects that cannot intersect or overlap with each other. To take this into account, non-crossing conditions can be imposed to constrain the geometry of networks, which consequently affects how they form, evolve and function. However, these constraints are not included in the theoretical frameworks that are currently used to characterize real networks5,6,7. Most tools for laying out networks are variants of the force-directed layout algorithm8,9—which assumes dimensionless nodes and links—and are therefore unable to reveal the geometry of densely packed physical networks. Here we develop a modelling framework that accounts for the physical sizes of nodes and links, allowing us to explore how non-crossing conditions affect the geometry of a network. For small link thicknesses, we observe a weakly interacting regime in which link crossings are avoided via local link rearrangements, without altering the overall geometry of the layout compared to the force-directed layout. Once the link thickness exceeds a threshold, a strongly interacting regime emerges in which multiple geometric quantities, such as the total link length and the link curvature, scale with the link thickness. We show that the crossover between the two regimes is driven by the non-crossing condition, which allows us to derive the transition point analytically and show that networks with large numbers of nodes will ultimately exist in the strongly interacting regime. We also find that networks in the weakly interacting regime display a solid-like response to stress, whereas in the strongly interacting regime they behave in a gel-like fashion. Networks in the weakly interacting regime are amenable to 3D printing and so can be used to visualize network geometry, and the strongly interacting regime provides insights into the scaling of the sizes of densely packed mammalian brains.

Network-based approach to prediction and population-based validation of in silico drug repurposing
Here we identify hundreds of new drug-disease associations for over 900 FDA-approved drugs by quantifying the network proximity of disease genes and drug targets in the human (protein–protein) interactome. We select four network-predicted associations to test their causal relationship using large healthcare databases with over 220 million patients and state-of-the-art pharmacoepidemiologic analyses.